Hemoglobinepathie
Screening
Door vroegtijdige diagnose kan gezondheidsschade als gevolg van complicaties, zoals sepsis, worden voorkomen door extra vaccinaties en profylactisch gebruik van antibiotica. Bij screening door middel van HPLC kunnen verschillende hemoglobinopathieën worden opgespoord.
Dragerschap diagnostiek is in ieder geval geïndiceerd bij:
• een microcytaire anemie zonder ijzergebrek
• bij aangetoond dragerschap is screening geïndiceerd bij partner en familie
• dragerschap bij een kind is een indicatie voor testen van de ouders
NB: Dragers van een hemoglobinopathie worden veelal gekenmerkt door een lichte verlaging van het Hb, echter een normaal Hb sluit dragerschap zeker niet uit. In geval van thalassemie is veelal sprake van een microcytair bloedbeeld, hetgeen differentiaal diagnostisch moet worden onderscheiden van ijzerdeficiëntie.
Bij zogeheten 'risico-paren' kan prenatale diagnostiek worden aangeboden in de vorm van een vlokkentest (10e - 12e wk) of amniocentese (16e wk).
Versie: december 2020
Thalassemie
Afhankelijk van de genetische mutatie kan thalassemie zich presenteren als symptoomloze laboratoriumafwijkingen t/m ernstige transfusie-afhankelijke anemie. Het gemeenschappelijke kenmerk is het microcytaire, hypochrome rode bloedbeeld.
Hemoglobinevarianten
Vele hemoglobinevarianten zijn het gevolg van single aminozuursubstituties in één van de globineketens. Er zijn meer dan 400 hemoglobinevarianten gedefinieerd, waarvan 50% klinisch geen betekenis hebben en de andere 50% veranderingen geeft in O2-affiniteit, of fysische instabiliteit van hemoglobine. De meest voorkomende en meest bekende hemoglobinevariant is HbS.
Sikkelcelziekte (SCD)
Sikkelcelziekte, één van de meest voorkomende erfelijke aandoeningen wereldwijd, is het gevolg van een puntmutatie in het β-globine gen (het gevolg van de substitutie van glutaminezuur door valine op positie 6 van de β-keten), waardoor hemoglobine S wordt gemaakt. Er zijn meerdere SCZ genotypes: homozygote type SCZ (HbSS) en compound heterozygote types SCZ (meest voorkomend zijn HbSC en HbSβ-thal, tevens HbSE, HbSOarab en HbSD). Polymerisatie van deoxy-HbS veroorzaakt hemolyse en vaso-occlusie met zeer diverse acute en chronische complicaties.
Er ontstaan veelal ziekteverschijnselen vanaf de zesde levensmaand, wanneer het HbF gehalte daalt. De klassieke symptomatologie bestaat uit recidiverende, pijnlijke vaso-occlusieve crises met orgaanschade in diverse organen. De ernst van de sikkelcelziekte is afhankelijk van meerdere factoren: de HbS concentratie, de mate van deoxygenatie en de hoeveelheid HbF. Daarnaast spelen diverse factoren in de microcirculatie een belangrijke rol: geactiveerde leukocyten, trombocyten, geactiveerde, beschadigde endotheelcellen en diverse cytokines en adhesiemoleculen.
Het beleid bij SCD wordt bepaald door:
1. Frequentie en ernst van acute crises
2. Chronische sequelae t.g.v. vaso-occlusieve crises, hemolyse en ijzerstapeling
De volgende crises worden beschreven:
1. klassieke vaso-occlusieve, pijnlijke crisis vaak met gegeneraliseerde microinfarcering, soms meer orgaan-gerelateerd
2. miltsequestratie en leversequestratie
3. aplastische crisis
4. hemolytische crisis
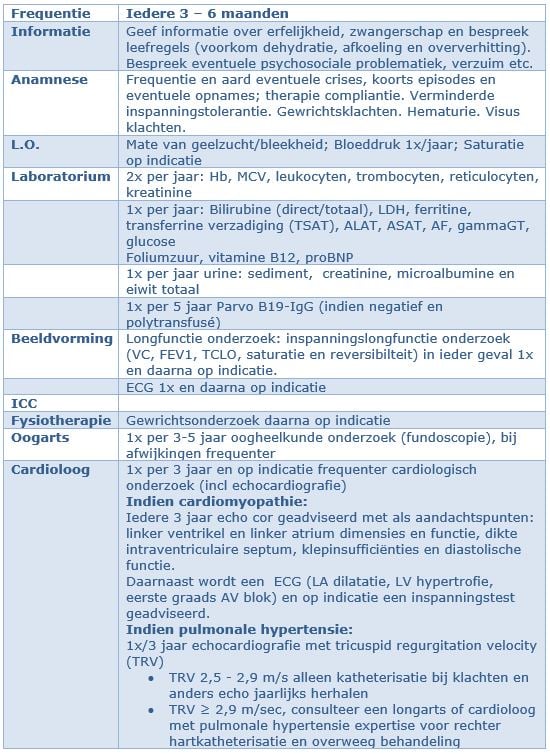
Complicaties SCD
Allogene stamceltransplantatie bij hemoglobinopathieen
Hoewel een allogene stamceltransplantatie vooralsnog de enige curatieve behandeling is, is er bij volwassenen slechts zeer beperkte ervaring.
Bij kinderen is er bij een allogene SCT met een MA conditonering een 5-jaarsoverleving en ziektevrije overleving van ongeveer 95%. De conditionering is echter toxisch, vooral bij patiënten ouder dan 16 jaar. Andere belangrijke problemen zijn het vinden van een HLA-identieke verwante/onverwante donor, allo-immunisatie t.g.v. frequente transfusies en reeds ontstane orgaanschade. Momenteel worden volwassen patiënten vooral in het AMC getransplanteerd. Er is structureel een MDO met het AMC waarbij potentiële kandidaten worden besproken. De beslissing om een patiënt te transplanteren wordt vooralsnog op individuele basis afgewogen. Indien tot transplantatie wordt besloten dient een uitgebreide inventarisatie te worden verricht van reeds bestaande orgaanschade. Gezien het relatief grote risico op morbiditeit en mortaliteit ligt een non-myeloablatief transplantatie regime bij volwassenen voor de hand.