Klinische Genetica
Patiëntenzorg is de kern van de bezigheden van de afdeling Klinische Genetica. Wij zien patiënten met vraagstellingen over erfelijkheid op de polikliniek, en verrichten genetische diagnostiek in ons laboratorium.
Voor patiënten
Een klinisch geneticus of andere klinisch-genetische zorgprofessional zoekt voor u uit of een aandoening erfelijk is. Er wordt onderzocht of er een verhoogde kans is voor uzelf of uw kinderen en zonodig wordt er DNA-diagnostiek aangevraagd. Zij bespreken de uitkomst van het onderzoek en de eventuele gevolgen voor u en uw familieleden.
In specifieke situaties bieden wij tevens psychologische begeleiding aan bij persoonlijke keuzes die u moet maken. Het kan zijn dat uw familieleden belang hebben bij de uitkomst van het erfelijkheidsonderzoek. In dat geval vragen wij u uw familie te informeren. Eventueel kunnen wij daarbij behulpzaam zijn.
Voor meer informatie over de polikliniek Klinische Genetica en de diverse spreekuren die wij doen kunt u hier klikken.
Voor aanvragers
De sectie Genoomdiagnostiek (GD) is verantwoordelijk voor de uitvoering en interpretatie van diagnostiek op het gebied van een groot aantal aandoeningen. Het GD is ISO15189 geaccrediteerd door de Raad voor Accreditatie onder nummer M007. De aanvraagformulieren en meer informatie over onze diagnostische werkzaamheden vindt u hieronder.
Voor meer informatie over het aanvragen van diagnostiek door niet-klinisch genetische zorgprofessionals verwijzen wij graag naar Leidraad aanvragen genetische diagnostiek in de kiembaan door niet klinisch genetische zorgprofessionals.pdf (vkgn.org) en de pagina Zelf diagnostiek aanvragen | Arts en Genetica .
Meer informatie over nevenbevindingen vindt u op Nevenbevindingen | Arts en Genetica
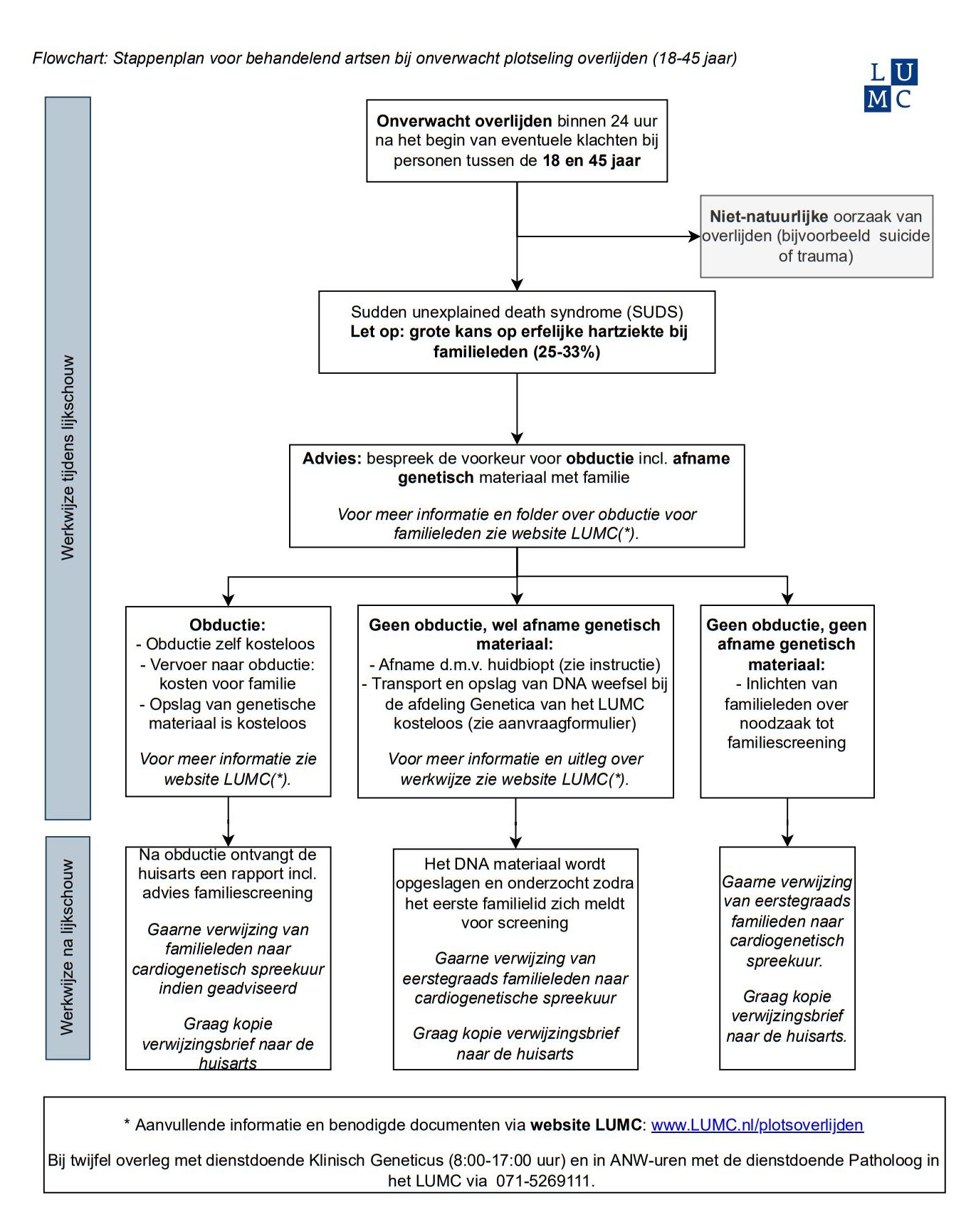
Plotse hartdood & genetica
In Nederland sterven er per jaar gemiddeld 16.000 mensen als gevolg van een acute en onverwachte hartstilstand. Dit betreft voor een belangrijk deel oudere patiënten, maar komt ook bij jongere personen voor. Extra alertheid is dan geboden. Wij adviseren obductie bij de overledene in de leeftijd tussen de 18-45 jaar.
Voor meer informatie zie Huisarts en Genetica.
De afdeling Klinische Genetica levert een actieve bijdrage aan de onderwijstaken van het LUMC op het vakgebied van erfelijkheidadvisering en klinisch genetische laboratorium diagnostiek. Dit bestaat uit onderwijs binnen het curriculum geneeskunde en biomedische wetenschappen (organisatie van blokken en lijnonderwijs, en het aanbieden van stageplaatsen), postacademisch onderwijs aan diverse specialismen en huisartsen.
Opleidingen
Clinical Genetics underlines the research goal of this centre which is: “To elucidate the molecular and clinical etiology of hereditary disease, congenital malformation, multifactorial disorders and hereditary cancer, in order to improve diagnostics, assist counselling, develop treatments and further prevention.”
Research programs
Contactgegevens
Polikliniek Klinische Genetica
Patiëntenzorgsecretariaat
Voor afspraken en vragen kunt u contact opnemen met:
Telefoonnummer: 071 526 80 33
E-mail: genetica@lumc.nl
Intercollegiaal overleg Artsenlijn
Telefoonnummer: 071 526 10 92
Laboratorium Genoomdiagnostiek/Monsterontvangst
Einthovenweg 20, 2333 ZC Leiden
Postbus 9600, 2300 RC Leiden
Gebouw 2, S6-P
Zorgverleners kunnen van maandag t/m vrijdag contact opnemen met:
Telefoonnummer: 071 526 98 00 bereikbaar van 8.30-12.30 en 13.30- 17.00 uur
E-mail: genoomdiagnostiek@lumc.nl
Stafsecretariaat
Telefoonnummer: 071 526 98 10
E-mail: stafsecretariaat.kg@lumc.nl
Kwaliteit en accreditatie
Accreditatie Laboratorium
De sectie Genoomdiagnostiek (GD) bestaat uit verschillende disciplines met een laboratoriumbreed kwaliteitszorgsysteem, gebaseerd op de internationale norm ISO 15189. De sectie GD met registratienummer M007 is sinds oktober 2006 geaccrediteerd door de RvA.
Accreditatie historie laboratorium
Moleculaire Genetica is sinds 1998 in het bezit van een RvA accreditatie, op basis van ISO 17025 (inclusief ISO 9001).
…Accreditatie Laboratorium
De sectie Genoomdiagnostiek (GD) bestaat uit verschillende disciplines met een laboratoriumbreed kwaliteitszorgsysteem, gebaseerd op de internationale norm ISO 15189. De sectie GD met registratienummer M007 is sinds oktober 2006 geaccrediteerd door de RvA.
Accreditatie historie laboratorium
Moleculaire Genetica is sinds 1998 in het bezit van een RvA accreditatie, op basis van ISO 17025 (inclusief ISO 9001).
Cytogenetica is in 2000 geaccrediteerd door zowel CCKL (Stichting voor de bevordering van de kwaliteit van het laboratoriumonderzoek en voor accreditatie van laboratoria in de gezondheidszorg) als de RvA. De CCKL erkenning is per 1 januari 2008 vrijwillig opgezegd. De reden van de opzegging was dat CCKL, in tegenstelling tot de RvA, geen internationale erkenning kan en mag afgeven.
Het Hemoglobinopathieën Laboratorium is sinds 2006 geaccrediteerd door de RvA.
&width=180&height=180)
&width=180&height=180)
&width=180&height=180)