Bottumor (botkanker)
Deze informatie is opgesteld door de afdeling(en) Orthopedie.
Wat is Bottumor (botkanker)?
Wat is een bottumor?
Bottumoren zijn zogenaamde sarcomen. Bij sarcomen gaat de tumor gaat uit van steunweefsel (botten, spieren, zenuwen en bloedvaten). Sarcomen zijn heel anders dan veel vaker voorkomende carcinomen: tumoren die ontstaan in organen zoals darm of borst.
Bij kwaadaardige tumoren is de celdeling niet meer onder controle en groeien de cellen overmatig. Kwaadaardige cellen vormen zo een gezwel (tumor) dat bestaat uit bijvoorbeeld bot-, kraakbeen- of bindweefselcellen. Soms zijn de cellen uit de tumor onder de microscoop nog goed te herkennen als bijvoorbeeld bot-, kraakbeen- of bindweefselcellen, maar soms lijken de cellen na het delen niet meer op de cellen waaruit ze zijn ontstaan. Dit noemen we atypie. De snelheid van tumorgroei en mate van atypie van de tumor cellen zegt iets over de mate van kwaadaardigheid van de tumor (gradering).
- Laaggradig: relatief langzaam delende cellen met een lagere kans op uitzaaiingen van de tumor op afstand (metastasen).
- Hooggradig: snel delende tumoren met veel atypische cellen en een hoge kans dat de tumor lokaal terug komt (recidief) of op uitzaaiingen van de tumor op afstand (metastasen).
- Intermediair: tussen laag- en hooggradig in.
Alleen bij kwaadaardige tumoren is er sprake van kanker. Kwaadaardige tumoren kunnen uitzaaiingen geven. Cellen uit een kwaadaardige tumor kunnen losraken van de tumor of een bloed- of lymfevat ingroeien en zich via de bloedbaan of de lymfevaten in het lichaam verspreiden. Zo kunnen ze ergens anders in het lichaam uitgroeien tot uitzaaiigen. Als er uitzaaiingen bij bot- en wekedelentumoren voorkomen, worden die meestal gevonden in de longen. Uitzaaiingen van deze tumoren kunnen ook voorkomen in het skelet, de buik, de lever of de lymfeklieren, maar dit komt minder vaak voor.
Welke soorten bottumoren zijn er?
Er zijn meerdere soorten bottumoren. De meest voorkomende zijn:
- een osteosarcoom: een kwaadaardige tumor die lijkt op bot;
- een Ewing-sarcoom: een kwaadaardige tumor die geheeld niet op normaal weefsel lijkt; komt voornamelijk voor in botweefsel, maar kan ook in de weke delen voorkomen;
- een chondrosarcoom: een kwaadaardige tumor die kraakbeen maakt.
- een reusceltumor: zeldzame lokaal agressief groeiende bottumor, waarbij het gezonde bot wordt afgebroken type reuscellen. Hieraan verleent de tumor ook zijn naam. Er bestaan ook een ander type reusceltumor die rondom pezen en in gewrichten groeit, maar dat is een andere soort tumor.
Waar komen bottumoren voor?
Bijna de helft van de bottumoren zit rond de heup of de knie. Ze kunnen ook voorkomen in de rest van het been, in de arm, het bekken of de wervelkolom. Heel soms zitten ze in handen of voeten, in de borstkas, het hoofd-halsgebied of de schedel. Als de tumor oppervlakkig ligt, wordt deze vaak sneller opgemerkt. Zit de tumor in het bekken of rond de wervelkolom, dan kan de tumor vaak ongemerkt groeien.
Sarcomen in de weke delen
Sarcomen zijn zeldzame en kwaadaardige tumoren die bestaan uit tumorcellen die lijken op cellen die we vinden in steunweefsel, zoals bot-, spier-, vet-, zenuw-, bloedvat- en bindweefsel. Op deze pagina’s vindt u uitgebreide informatie over sarcomen in botweefsel. Wilt u meer weten over sarcomen in de overige weefsels, de zogeheten weke delen? Op onze pagina over wekedelentumoren informeren we u verder.
Verschijnselen
De klachten bij een kwaadaardige bottumor zijn in eerste instantie vaak vaag. Vaak is pijn is de eerste klacht. Meestal is er ook s’ nachts pijn. De pijn zit vaak in of rond het bot of het naastgelegen gewricht. Soms treedt er een zwelling op en wanneer dit dicht bij een gewricht is, kan bewegingsbeperking ontstaan. Ook kan er een spontane botbreuk optreden door de aantasting van het bot.. Bottumoren net onder de huid vallen weliswaar beter op, maar veroorzaken vaak weinig klachten. Bovendien kunnen ze lijken op onschuldige knobbeltjes, zoals vetbulten.
Oorzaak
Bottumoren zoals het osteosarcoom en het Ewing sarcoom komen met name voor bij kinderen en jong volwassenen. Het chondrosarcoom kan op alle leeftijden voorkomen, maar meestal pas na het 30ste levensjaar. Meestal is er geen directe oorzaak voor het ontstaan van deze tumoren aan te wijzen. Heel soms is er sprake van een familiaire belasting, waardoor tumoren vaker voorkomen in bepaalde families.
Waarom u bij ons in goede handen bent
Sneldiagnose sarcoom
Mede dankzij onze ruime ervaring met sarcomen kunt u bij ons terecht voor sneldiagnose. Meestal hebt u al binnen 3 dagen na een verwijzing een afspraak met een van onze specialisten. We plannen onderzoeken zo veel mogelijk op dezelfde dag en een week later hoort u de uitslag. Zo zorgen we dat u zo snel mogelijk weet waar u aan toe bent.
Multidisciplinair team
Zowel tijdens de onderzoeken als de behandeling bent u in handen van een multidisciplinair team. Specialisten van verschillende afdelingen dragen er samen zorg voor dat u de behandeling krijgt die het beste bij u past.
Ondersteuning voor jongvolwassenen met kanker
In het LUMC is een speciaal AYA-team dat ondersteuning biedt aan adolescenten (A) en jongvolwassenen (young adults; YA) in de leeftijd van 16 tot 35 jaar die kanker hebben of hebben gehad. Bij dit team kunnen zij terecht met vragen over de ziekte, maar bijvoorbeeld ook over studie, werk, kinderwens, hypotheken, relaties en seks.
Wetenschappelijk onderzoek naar sarcomen
Het LUMC heeft erg veel kennis over sarcomen in huis en breidt die kennis ook steeds verder uit. We doen veel wetenschappelijk onderzoek naar sarcomen en zijn constant op zoek naar nog betere behandelingen en methoden voor diagnostiek. Vaak zijn die onderzoeken groots opgezet in samenwerking met vooraanstaande ziekenhuizen uit de hele wereld. Soms kunt u ook deelnemen aan zo’n studie. We noemen dat clinical trials. U wordt dan bijvoorbeeld behandeld met medicijnen die ergens anders nog niet verkrijgbaar zijn. Dit kan een positieve bijdrage leveren aan uw behandeling. Als er een clinical trial loopt, doet het LUMC daar meestal aan mee. Zowel bij nationale als internationale studies.
Steun ons onderzoek naar botsarcoom
Binnen het LUMC werkt een groot multidisciplinair team samen aan een breed onderzoek om botsarcoom en de gevolgen hiervan beter te kunnen behandelen. Dit onderzoek moet bijdragen een betere kwaliteit van leven voor de jongeren die deze ziekte krijgen en een grotere kans op genezing.
U kunt daarbij helpen! Steun ons onderzoek naar botsarcoom.
Als bij u de verdenking bestaat op een kwaadaardige bottumor, dan kunt u bij het LUMC terecht voor sneldiagnostiek. Uw huisarts of specialist verwijst u daarvoor door naar de polikliniek Orthopedie. Meestal kunt u binnen enkele werkdagen terecht bij 1 van onze medisch specialisten. Die zorgt meteen dat u de juiste onderzoeken krijgt en een week later hebt u al een uitslag.
Afspraak maken voor sneldiagnose botkanker
U hoeft niet zelf een afspraak te maken voor sneldiagnose. Uw huisarts of verwijzer meldt u aan via de polikliniek Orthopedie. De secretaresse van onze polikliniek belt u daarna voor het maken van een afspraak. U kunt meestal binnen enkele werkdagen bij ons terecht voor een (telefonisch) consult met een medisch of verpleegkundig specialist.
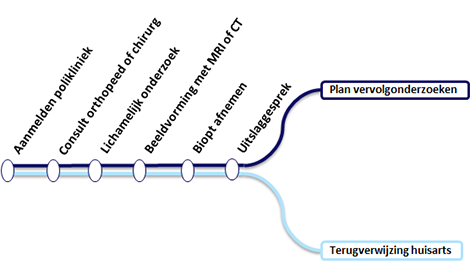
Schematische weergave van alle stappen bij sneldiagnose botkanker.
Op de dag van de afspraak zelf kunt u zich melden aan de balie van de polikliniek Orthopedie. Deze bevindt zich op in het polikliniekgebouw Poligebouw Heelkunde, Orthopedie en Gipskamer (routenummer 1000) naast de parkeergarage van het LUMC.
Wie komt u tegen en wat kunt u verwachten?
U hebt een gesprek met een oncologisch orthopedisch chirurg of met de arts in opleiding tot specialist. U krijgt een lichamelijk onderzoek en we kijken welk onderzoek nog meer nodig is voor een goede diagnose. Als het mogelijk is, voeren we die onderzoeken op dezelfde dag nog uit. U hoeft dan niet nog een keer langs te komen. Lukt dit niet, dan zorgen we dat u binnen een week na het gesprek terecht kunt.
Mogelijke onderzoeken
- Vaak wordt er als eerste een röntgenfoto van de aangedane plek of zwelling gemaakt. Hierbij kunnen afwijkingen in het bot goed worden weergegeven. Met een röntgenfoto is het soms al mogelijk onderscheid te maken tussen goed- en kwaadaardige tumoren van het bot. Wanneer er een verdenking op een kwaadaardige tumor is, maken we aanvullend vaak een MRI of CT-scan.
- Een MRI-scan vertelt ons exact de plaats van de tumor. Bij een MRI-onderzoek komt er veel geluid uit het apparaat. Daarom heeft u een koptelefoon op en kan u tijdens het onderzoek via de intercom contact houden met de radiologisch laborant. Bij dit onderzoek krijgt u via een infuus vaak een contrastmiddel ingespoten.
- Een CT-scan zegt veel over de kwaliteit van het omliggende bot. Bij een CT-scan ligt u op een beweegbare tafel die langzaam door het apparaat heen schuift. Soms is voor het maken van deze foto’s contrastvloeistof nodig. Dit wordt dan in een bloedvat van uw arm gespoten.
- Bloedonderzoek.
- Een botscan of PET/CT-scan om te kijken of er uitzaaiingen zijn. Deze scan wordt gemaakt met een kleine hoeveelheid radioactieve stof. Na het onderzoek plast u deze stof weer uit. U ligt voor dit onderzoek op een tafel en merkt niets van het maken van de beelden.
- CT-scan van de longen. Deze scan wordt gemaakt om te kijken of er uitzaaiingen in de longen zijn.
- Het wegnemen van een stukje weefsel uit het bot (biopsie). Hiermee kan worden bepaald om wat voor afwijkend weefsel het gaat. Een patholoog die zich heeft gespecialiseerd in bot- en wekedelentumoren onderzoekt het weefsel.
- Wanneer een of meerdere van deze onderzoeken al in een ander ziekenhuis bij u gebeurd zijn, zullen wij de onderzoeken opvragen en opnieuw beoordelen. Het zelfde onderzoek in het LUMC herhalen is dan vaak niet nodig.
Wel of geen biopsie?
Een biopsie geeft definitief uitsluitsel over de tumor. Een biopsie is echter niet altijd nodig. Soms blijkt al uit de röntgenfoto of de MRI-scan wat de diagnose is.
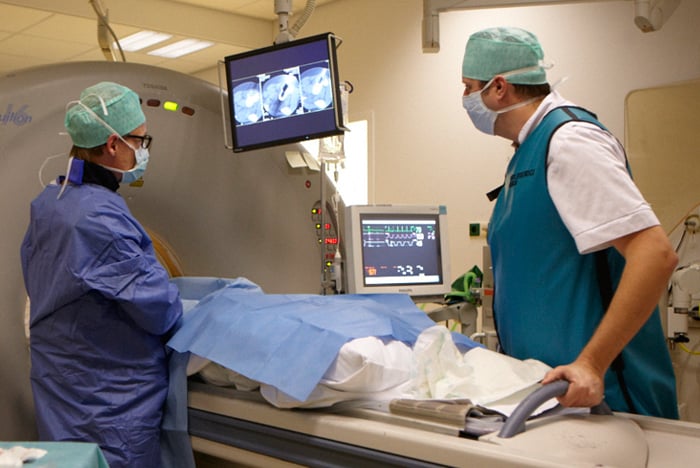
De interventieradioloog haalt tijdens een biopsie een klein stukje weefsel weg bij een patiënt.
Uitslag van het onderzoek
Binnen 2 weken hebt u opnieuw een afspraak, ditmaal met de orthopeed of chirurg en een verpleegkundig specialist. Dit gebeurt meestal nadat de uitslagen van uw onderzoek zijn besproken in het multidisciplinair overleg. Dit overleg vindt elke vrijdagmorgen plaats tussen medisch specialisten onderling. De uitslagen worden in het consult met u besproken en u krijgt informatie over een eventueel behandelplan.
Welke behandelingen zijn er mogelijk?
Iedere bottumor gedraagt zich anders. Daarom bepaalt uw arts samen met het multidisciplinaire team voor u een behandeling op maat. We kijken hoe we de tumor het beste kunnen behandelen, maar houden ook rekening met uw persoonlijke wensen. Behandelkeuzes maken we altijd in overleg met u, ook omdat de behandeling ingrijpend kan zijn.
Overleg tussen patiënt en arts
Bij een behandeling richten we ons op het veilig verwijderen van de tumor, met behoud van zo veel mogelijk functie en beweeglijkheid. Uw orthopedisch chirurg bespreekt met u en uw naasten het behandeladvies dat het medisch team heeft opgesteld. De voor- en nadelen van alle behandelmogelijkheden komen daarbij aan de orde. We proberen de behandeling altijd zo veel mogelijk af te stemmen op uw persoonlijke omstandigheden. Als u bijvoorbeeld na een operatie wilt kunnen blijven sporten, dan houden wij daar zo veel mogelijk rekening mee.
Welke behandelingen zijn er?
Voor bottumoren bestaan 3 hoofdbehandelingen:
- Operatieve verwijdering van de tumor
- Bestraling
- Chemotherapie
Vaak bestaat de behandeling uit een combinatie van de 3. De volgorde kan wisselen.
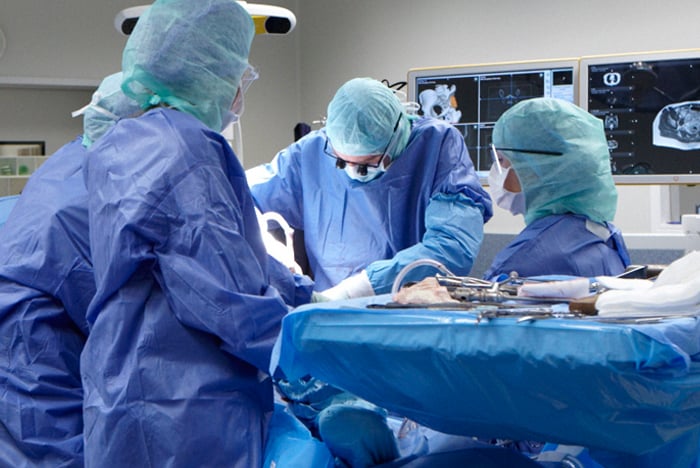
Een chirurgisch team in de operatiekamer behandelt een patiënt aan een bottumor.
Operatieve verwijdering van de tumor
Bij een operatie nemen we de tumor in het algemeen zo ruim mogelijk weg. Dit betekent dat we proberen een laagje gezond weefsel rondom de tumor te laten zitten, zodat de kans dat de tumor terugkomt zo klein mogelijk wordt. In het LUMC hebben we veel ervaring met een zo klein mogelijke grens (marge) tussen de tumor en het gezonde weefsel te laten om een goed functioneel resultaat te kunnen bieden en de kansen op overleving zo groot mogelijk te houden. Hiervoor zijn verschillende technieken ontwikkeld zoals computernavigatie, fluorescentie chirurgie en pre-operatieve radiotherapie.
Soms is het nodig een deel van het aangetaste bot te verwijderen. Zo’n operatie proberen we altijd zo beperkt mogelijk te houden. Want hoe kleiner de ingreep, des te minder verlies van functie en beweeglijkheid, en des te kleiner de kans dat er complicaties optreden. In enkele gevallen is het nodig om het hele bot te verwijderen. Bij een goedaardige tumor wordt doorgaans minder bot verwijderd dan bij een kwaadaardige tumor. Bij een goedaardige tumor is chemotherapie of bestraling bijna nooit nodig. Soms is het zelfs helemaal niet nodig een goedaardig gezwel te behandelen. In het geval van een operatie brengt u eerst een bezoek aan de anesthesist. Met hem of haar bespreekt u de pijnstilling en de narcose.
Na de operatie bestudeert een patholoog het weggenomen weefsel. Soms vind de patholoog toch nog levende tumorcellen in de rand (grens) van het weefsel. Het is dan soms noodzakelijk opnieuw een operatie uit te voeren, waarbij we nog meer weefsel weghalen. Hierna volgt dan in veel gevallen radiotherapie (bestraling).
Bot vervangen
Het verwijderde botdeel kan vaak vervangen worden. Dit kan bijvoorbeeld met een:
- gewrichts- of botprothese;
- Donorbot (dit is afkomstig van een botbank);
- bottransplantatie, bijvoorbeeld van uw kuitbeen naar het verwijderde deel van het bot;
- combinatie van bovenstaande.
Kunstgewricht en amputatie
Als een tumor dicht bij een gewricht zit, is het niet altijd mogelijk het gewricht te behouden. Een kunstgewricht kan dan een oplossing zijn. Wanneer het niet mogelijk is de tumor veilig te verwijderen zonder de functie van een ledemaat te verliezen, dan is een amputatie soms het enige alternatief. Dit komt bij ongeveer 1 op de 15 patiënten voor.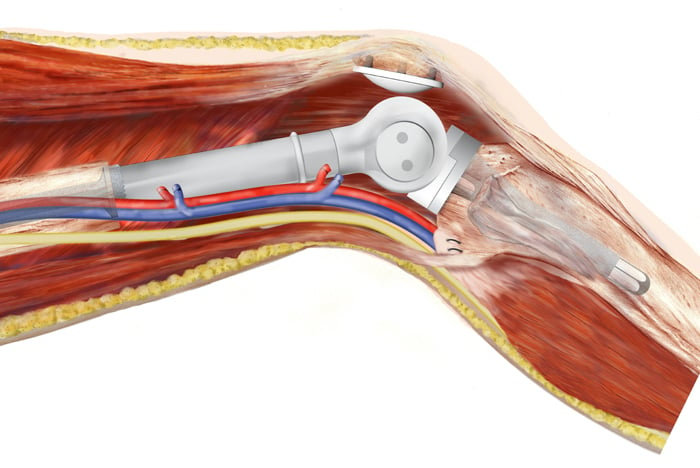
Een illustratie toont goed hoe een knieprothese functioneert.
Chemotherapie en bestraling
Een operatie is vrijwel altijd noodzakelijk bij de behandeling van een bottumor. Of u daarnaast ook chemotherapie krijgt en/of bestraald wordt, hangt vooral af van het soort bottumor dat u hebt. Soms is het beter om eerst te zorgen dat eventuele niet-waarneembare of bewezen uitzaaiingen (metastasen) worden behandeld of voorkomen met chemotherapie.
Bij een Ewing- en bij osteosarcoom krijgt u eerst chemotherapie en/of radiotherapie. Dit heet neo-adjuvante chemotherapie. Als de chemotherapie wordt gegeven na de operatie noemen we dit adjuvante chemotherapie. Deze (neo)adjuvante therapie is er op gericht om uw prognose te verbeteren.
Bij een chondrosarcoom opereren we meestal direct, zonder voorafgaande chemotherapie of bestraling. Want een chondrosarcoom is vrij ongevoelig voor chemo- of radiotherapie.
Botkanker bij kinderen
AYA-patiënten en volwassenen met botkanker worden behandeld in het LUMC. Ook kinderen met botkanker worden behandeld in het Prinses Maxima Kinderoncologisch Centrum (PMC) in Utrecht. Mocht er bij uw kind een verdenking zijn op een kwaadaardige bottumor, dus een vorm van botkanker, dan kunnen de onderzoeken die noodzakelijk zijn voor het stellen van de diagnose kunnen in het LUMC worden verricht. Indien de diagnose van een kwaadaardige bottumor bevestigd wordt zal uw kind direct worden doorverwezen voor behandeling in het PMC. Vanuit het LUMC werkt Prof. dr. M.A.J. van de Sande ook gedeeltelijk in het PMC.
Hoe kunt u zich op de behandeling voorbereiden?
Alle onderdelen van de behandeling kunnen ingrijpend zijn. Daarom is het goed dat u zich oriënteert. Wat houdt de ziekte in? Wat zijn de behandelmogelijkheden? U kunt erover lezen en erover praten met uw familie en vrienden.
Vragen kunt u altijd stellen in het gesprek met uw behandelaar. U kunt ook bellen als er thuis vragen boven komen. De verpleegkundige is bereikbaar via de polikliniek Orthopedie: 071 - 526 80 03.
Wat is de prognose?
Omdat kwaadaardige bottumoren onderling zeer verschillend zijn, is het moeilijk een algemene prognose te geven. De behandeling is er in eerste instantie op gericht om u te genezen. Dit betekent vaak dat we de tumor moeten verwijderen en moeten voorkomen dat de tumor terug komt. Dit heet een curatieve behandeling. Soms kan de tumor niet meer worden verwijderd, omdat deze te dicht bij belangrijke organen ligt of dat deze al is uitgezaaid naar plekken in de longen en/of andere organen.
Landelijk gezien is 60 procent van de patiënten 5 jaar na de behandeling nog in leven. Maar dit varieert erg per tumor type. Het LUMC doet er alles aan dit overlevingspercentage omhoog te krijgen. Dat is de reden dat we meedoen met een groot aantal internationale onderzoeken. Wij stellen het zeer op prijs als u daaraan deelneemt.
Meedoen aan wetenschappelijk onderzoek
Het LUMC is niet alleen een centrum van geavanceerde medische zorg, maar ook van geavanceerd medisch onderzoek. Dit houdt in dat u kunt meedoen aan de nieuwste studies, met de nieuwste medicijnen. Mogelijk hebben deze bij u meer effect dan de standaardbehandeling. De oncoloog kan u vertellen aan welke onderzoeken u kunt meedoen. De orthopedisch chirurg zal u naar de oncoloog verwijzen.
Wetenschappelijk onderzoek naar sarcomen
Het LUMC heeft erg veel kennis over sarcomen in huis en breidt die kennis ook steeds verder uit. We doen veel wetenschappelijk onderzoek naar sarcomen en zijn constant op zoek naar nog betere behandelingen en methoden voor diagnostiek. Vaak zijn die onderzoeken groots opgezet in samenwerking met vooraanstaande ziekenhuizen uit de hele wereld. Soms kunt u ook deelnemen aan zo’n studie. We noemen dat clinical trials. U wordt dan bijvoorbeeld behandeld met medicijnen die ergens anders nog niet verkrijgbaar zijn. Dit kan een positieve bijdrage leveren aan uw behandeling. Als er een clinical trial loopt, doet het LUMC daar meestal aan mee. Zowel bij nationale als internationale studies.
Bij een bottumor is een operatie of bestraling meestal niet het einde van de behandeling. Zo kan het zijn dat u revalidatie nodig hebt. Ook daar helpen we u bij. Verder is het belangrijk dat u alert blijft op alarmsymptomen. We kunnen u dan bij onverwachte gevolgen zo snel mogelijk helpen.
Welke specifieke nazorg kunnen we bieden bij deze aandoening?
Nazorg is bijna altijd nodig, zeker bij het plaatsen van een prothese en bij een amputatie. Revalidatie is vaak een onderdeel van het herstel. Ook kan het zijn dat u graag hulp ontvangt bij het verwerken van de ingreep. In dat geval kunt u contact opnemen met een oncologisch maatschappelijk werker.
Revalidatie
De afdelingen Revalidatiegeneeskunde en Fysiotherapie van het LUMC hebben veel ervaring met de nabehandeling van bottumoren. Onze revalidatiearts beoordeelt of nabehandeling in een revalidatiecentrum noodzakelijk is, of dat u thuis kunt revalideren of samen met een fysiotherapeut. Wij zorgen ervoor dat deze zorg wordt geleverd, zowel binnen het LUMC als bij u in de buurt.

Verzameling attributen in de sportzaal van de afdeling Fysiotherapie.
Psychologie en maatschappelijk werk
De behandeling van bottumoren is vaak heel ingrijpend, voor u als patiënt, maar ook voor uw naasten. Het LUMC biedt u ook ondersteuning op het gebied van psychologische zorg en maatschappelijk werk.
Behandelteam
Bij de diagnostiek en behandeling van een bottumor is een groot aantal specialisten van het LUMC betrokken. Samen beoordelen ze uw situatie en zoeken ze naar de best mogelijke behandeling. In dit multidisciplinaire team treft u onder meer een oncoloog, een radioloog, een radiotherapeut, een interventieradioloog, een neurochirurg, een plastisch chirurg en een patholoog. Sommige van deze specialisten zult u ontmoeten, anderen zetten zich vooral achter de schermen voor u in.
Bij de diagnostiek en behandeling van een sarcoom is een groot aantal specialisten van het LUMC betrokken. Samen beoordelen zij uw situatie en bepalen de best mogelijke behandeling. In dit multidisciplinaire team treft u onder meer een orthopeed, chirurg, oncoloog, radioloog, radiotherapeut, interventieradioloog, neurochirurg, plastisch chirurg en een patholoog. Sommige van deze specialisten zult u ontmoeten, anderen zetten zich vooral achter de schermen voor u in.

Dr. R.J.P. van der Wal
Oncologisch orthopedisch chirurg

Drs. D. Broekhuis
Oncologisch orthopedisch chirurg

Prof. dr. M.A.J. van de Sande
Oncologisch orthopedisch chirurg

M. van Duijvenbode
Verpleegkundig Specialist

N.A.C. Leijerzapf
Verpleegkundig specialist

Prof. dr. J.V.M.G. Bovée
Patholoog

S.W. Lam
Patholoog

Prof. dr. P.C.W. Hogendoorn
Patholoog
Voorzitter Nederlandse Commissie voor Beentumoren

Prof. A.J. Gelderblom
Oncoloog

Dr. F.M. Speetjens
Medisch oncoloog

M. Nieuwenhuis
Verpleegkundig specialist

Dr. M.C. Burgmans
Interventieradioloog

Dr. A.R. van Erkel
Interventieradioloog

Dr. C.S.P. van Rijswijk
Interventieradioloog

Dr. R.W. van der Meer
Interventieradioloog

Prof. dr. L.F. De Geus-Oei
Nucleair geneeskundige

Dr. D. Vriens
Nucleair geneeskundige

Prof. Dr. J.A. van der Hage
Oncologisch chirurg

Dr. H.H. Hartgrink
Oncologisch chirurg
Dr. G.J. Liefers
Oncologisch chirurg

Drs. A. Navas Cañete
Radioloog

Dr. M. Reijnierse
Radioloog

Dr. K. van Langevelde
Radioloog

Prof. dr. W.C. Peul
Neurochirurg
Afdelingshoofd Neurochirurgie

Dr. Dr. W. Pondaag
Neurochirurg

Dr. C.L.A. Vleggeert-Lankamp
Neurochirurg

Dr. J.L. Groen
Neurochirurg

Drs. P.J. Schutte
Neurochirurg

Dr. R.L.M. Haas
Radiotherapeut-Oncoloog

Dr. L.M. Wiltink
Radiotherapeut-Oncoloog

Drs. R.G. Moeri-Schimmel
Radiotherapeut-oncoloog

Dr. A.D.G. Krol
Radiotherapeut-oncoloog
Wie kunt u nog meer tegenkomen?
- Plastisch chirurgen: Drs. L.U.M. Corion, Drs. G.K. van Drunen, Drs P.S. Verduijn
- Fysiotherapeuten
- Psychologen
- Ergotherapeuten
- Maatschappelijk medewerkers
Aan welke studies kan je meedoen?
Patiëntgebonden onderzoek
Onze specialisten doen veel onderzoek naar de biologische achtergronden van bottumoren en andere ziekten. Kennis over deze aandoeningen kan alleen maar groeien als patiënten deelnemen aan dit onderzoek. We stellen het daarom zeer op prijs als u hiertoe bereid bent. Deze patiëntgebonden studies richten zich op verbetering van de eerste behandeling van bottumoren en de ontwikkeling van nieuwe behandelingen voor patiënten waar geen antitumor-opties meer voor zijn. Binnen het LUMC lopen meerdere studies, die regelmatig wisselen.
Gebruik van nieuwe geneesmiddelen
Als patiënt hebt u er ook profijt van dat het LUMC naast een zorgcentrum een onderzoekscentrum is. Zo worden bij ons de nieuwste geneesmiddelen getest. Als de standaardmedicijnen bij u onvoldoende werken, heeft een experimenteel middel mogelijk meer effect. Deze middelen worden op het LUMC in studieverband aangeboden.
Als uw situatie zich hiervoor leent en u voldoet aan de criteria, kunt u meedoen met deze clinical trials. U kunt uw arts hiernaar vragen, maar het behandelteam zal ook zelf alle behandelmogelijkheden uitgebreid met u bespreken.

Een onderzoeker bekijkt weefsel onder een microscoop.
Contact
Wilt u meer weten of hebt u nog vragen? Neem dan contact met ons op of volg de links voor aanvullende informatie.
Patiëntportaal mijnLUMC
In het patiëntportaal mijnLUMC vindt u een duidelijk overzicht van uw behandelingen en hebt u inzicht in uw medische gegevens. Snel en veilig. Thuis, onderweg en in het ziekenhuis.
Patiënt verwijzen
Informatie voor artsen en instellingen die patiënten naar het LUMC willen verwijzen.
Contactgegevens voor patiënten
- Polikliniek Orthopedie: 071 - 526 80 03
- Polikliniek Radiotherapie: 071 - 526 35 25
- Polikliniek Medische Oncologie: 071 - 526 35 23
Overig
Verwijsinformatie voor huisartsen en andere medisch specialisten
Spoed / Sneldiagnostiek
Bij spoed, verdenking op een maligne tumor of twijfel over de aard van de tumor kunt u contact opnemen met:
- Mw. N.A.C. Leyerzapf en Mw. M. van Duijvenbode, verpleegkundig specialisten
- E-mail: sarcomen@lumc.nl
- Telefoonnummer: 071 - 526 20 60
In de regel kunnen patiënten binnen 1 week gezien worden op de polikliniek. Eventueel kunt u voor overleg contact opnemen met een van de betrokken specialisten via onderstaande contactgegevens.
Contactgegevens betrokken afdelingen
Alle overige patiënten kunt u schriftelijk aanmelden door middel van een verwijsbrief gericht aan de betrokken afdeling:
Polisecretariaat Orthopedie
- Bereikbaar tussen 09.00 en 12.30 uur
- Tel: 071 - 526 80 03
- Email: polikliniekorthopedie@lumc.nl
Polisecretariaat Heelkunde
- Bereikbaar tussen 8.30 en 16.00 uur
- Tel: 071 - 526 36 04
Secretariaat Oncologie
- Bereikbaar tussen 8.30 en 16.00 uur
- Tel: 071 - 526 34 64 of 071 - 526 34 86
- Email: medischeoncologie-extern@lumc.nl of oncology@lumc.nl
Secretariaat Radiotherapie
- Bereikbaar tussen 8.30 en 16.00 uur
- Tel: 071 - 526 35 25
Verwijzing van kinderen met een bottumor
Benigne bottumoren:
Kinderen met (een verdenking op) een benigne bottumor kunt u verwijzen naar de specialisten van één van de expertisecentra in Nederland.
Voor het Leids Universitair Medisch Centrum zijn dat Prof.Dr. M.A.J. van de Sande, Dr. R.J.P. van der Wal en drs. B. Broekhuis.
Maligne bottumoren:
Kinderen met een (hoge) verdenking op een maligne bottumor kunnen worden verwezen naar de orthopedisch oncologen verbonden aan één van de expertisecentra of aan het Prinses Maxima Centrum in Utrecht. Voor het LUMC is dat Prof. Dr. M.A.J. van de Sande.
In geval van twijfel met betrekking tot benigniteit of maligniteit is het verstandig te overleggen met één van de orthopedisch oncologen verbonden aan één van de expertise centra.
Multidisciplinair overleg sarcomen
Iedere vrijdagochtend worden patiënten met botsarcomen en wekedelensarcomen in het LUMC besproken in een multidisciplinair overleg. Alle betrokken specialismen zijn hierbij aanwezig. Voor ondersteuning in het diagnostisch of behandeltraject is het mogelijk om patiënten van buiten het LUMC aan te melden voor deze bespreking.
U kunt patiënten aanmelden per e-mail (sarcomen@lumc.nl) of via telefoonnummer 071 - 526 20 60. Ook kunt u voor overleg contact opnemen met een van de betrokken specialisten via bovenstaande contactgegevens.
Voor deze bespreking hebben wij minimaal de volgende informatie nodig:
- De persoonsgegevens van de patiënt (inclusief telefoonnummer).
- Naam van de verwijzer.
- Vraagstelling.
- Alle beeldvorming en bijbehorende verslagen. Deze kunt u opsturen naar de poli Orthopedie, t.a.v. Mw. N.A.C. Leyerzapf, postbus 9600, 2300 RC Leiden.
- Alle coupes en weefselbiopten. Indien aanwezig opsturen naar de afdeling Pathologie.
Patiënten die u voor woensdag 12.00 uur aanmeldt, worden diezelfde vrijdag besproken. Hiervoor dienen de benodigde beeldvorming en histologie uiterlijk woensdag binnen te zijn.
Verslaglegging
De inzender wordt schriftelijk op de hoogte gesteld van de (differentiaal) diagnose en het eventuele advies zoals vastgesteld tijdens de bespreking.
Behandelrichtlijnen
Beentumorencommissie
Voor een onafhankelijk advies over diagnostiek en/of behandeling kunt contact opnemen met de Nederlandse Commissie voor Beentumoren.
Adres
Commissie voor Beentumoren
p.a. Leids Universitair Medisch Centrum
Postzone C-2-S
Postbus 9600, 2300 RC Leiden
Telefoon: 071-5263490
Email: cvb@lumc.nl
Website
https://commissievoorbeentumoren.nl
De Commissie heeft als primaire taak een, (inter)nationaal erkend, consulentschap ter medebeoordeling van patiënten met tumoren of daarop lijkende afwijkingen van het skelet en wekedelen. Tot op heden zijn geen kosten aan consultatie van de Commissie verbonden. Daarentegen kunnen geen kosten, bijvoorbeeld voor verzending of kopiëren, bij de Commissie in rekening gebracht worden. De Commissie bestaat sinds 1953.
Procedure reguliere consulten
Voor een correcte beoordeling van de casus moet de Nederlandse Commissie voor Beentumoren kunnen beschikken over volledige informatie:
- Klinische gegevens (anamnese, aard en duur van de klachten, bevindingen bij onderzoek, laboratoriumgegevens, operatieverslag).
- Alle relevante röntgenonderzoeken incl. CT-, MRI- en isotopenonderzoeken voor zover verricht en alle verslagen van deze onderzoeken.